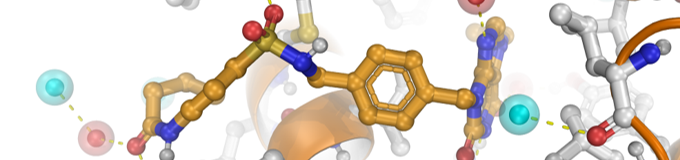
 ChemoDOTS Overview
ChemoDOTS OverviewThis web server is designed to build focused chemical libraries, starting from a user-specified hit fragment, using chemistry-driven approach and according to the growing strategy encountered in Fragment-Based Drug Discovery. This workflow represents the initial in silico steps of our DOTS (Diversity-Oriented Target-focused Synthesis) methodology (Hoffer et al., 2018) that combines computer-based library design and virtual screening with automated chemical synthesis and compound evaluation.
Typically, a focused chemical library is generated by combining an activated core analog of a hit fragment, with a collection of functionalized building blocks using medicinal chemistry-relevant in silico encoded chemical reactions. This last point guarantees that the designed compounds can be easily synthesized in one or two steps. Physicochemical filters can then be applied to extract a diverse subset of representative compounds that possess reasonable properties. Finally, a "ready-to-dock" virtual library is provided in the widely accepted mol2 format that is compatible with most docking programs.
The ChemoDOTS web server generates a diverse set of H2L candidate molecules while maximizing synthetic feasibility, helping scientists more rapidly and efficiently achieve H2L optimization goals for drug development.
Generate Focused Virtual Libraries for Your Activated Hit Fragment Start the ChemoDOTS web server here.
How Does it Work
Start the ChemoDOTS web server here.
How Does it WorkTypically, an activated form of the reference fragment is virtually coupled to a database of up-to-date commercially available building blocks using an ensemble of encoded chemical reactions encountered during the hit-to-lead (H2L) stages in the pharmaceutical industry (Hartenfeller et al., 2011). One of the main advantages of the proposed approach is to consider the synthetic accessibility of the designed compounds (Hoffer et al., 2018b). All compounds proposed in the final chemical library can be synthesized efficiently in just one or two steps with high yields.
A customizable postprocessing stage (Filtering) is then applied to extract a subset of representative and clean compounds that also possess reasonable computed physico-chemical properties. At this stage different properties can be fine-tuned (such as Molecular Weight, LogP, Hydrogen Bond Donors, Hydrogen Bond Acceptors, Fsp3). In the final post-processing stage, the following steps are executed: explicit stereoisomer computation, generation of a single 3D conformer for each compound and the creation of a 'ready-to-dock' file in mol2 format.
Chemical libraries are generated starting from a single user-defined structure as follows:
 Important note
Important note
The reference molecule needs to contain at least one of the following functions: Acid; Alcohol; Aldehyde; Alkene; Alkyne; Amidine; Amine; Boronate; Carboxylic Acid; Dicarbonyl; Ester; Hydrazine; Isocyanate; Isothiocyanate; Ketone; Nitrile; Organo Halide; Stannane; Sulfonylhalide; Sulfonamide; Thioamide; Thiol. (see exemples here).
An optional post-processing stage is available:
All generated files can be downloaded via an archive that contains:
 References
References
